

It was first announced four years ago. Delayed for a year due to the COVID-19 pandemic. Every medical device manufacturer selling to the European Union or hoping to has been preparing and awaiting its arrival with a combination of anticipation and dread. The EU Medical Device Regulation (MDR) is set to take effect on May 27, 2021. Failure to provide the proper compliance documentation could mean fines, litigation, product recalls, design changes, and even losing access to sizable market opportunities.
The EU MDR replaces the EU Medical Device Directive (MDD), and the replacement of the word “directive” with “regulation” tells the story. The new laws are more stringent, reflecting the demand within the European Union for greater protection of health and privacy. The EU MDR demands a more transparent and proactive approach to addressing medical device products that could negatively impact human health and the environment. And its regulations extend well beyond the pre-approval stage that was the focus of the previous directives; instead, the new regulations take a full product life cycle approach.
The new laws also broaden the universe of those products that need to be MDR-certified. Now, it’s not just purely medical products that are targeted. Products formerly considered cosmetic have been added to the list of regulated items, including cosmetic contact lenses, facial and other subcutaneous fillers, and tattoo and hair removal equipment. Requirements vary depending on intended use, targeted contact with the patient and duration.
Does Europe Really Matter?
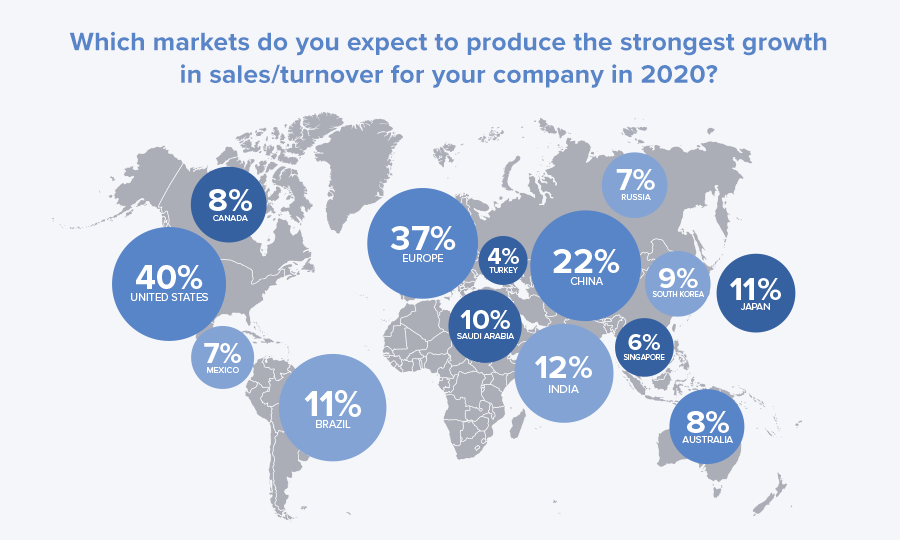
Yes! With a market estimate of $147 billion in 2018 (122 billion Euros), there’s no denying Europe is a sizable opportunity for medical device manufacturers, accounting for nearly one-third of the global market. A recent Emergo Outlook survey asked a similar question to manufacturing leaders regarding future sales (see figure below).
New MDR Definitions for “Medical Device”
The MDR expands the definition of the term “medical device” as an instrument, apparatus, appliance, software, implant, reagent, material, or other article” that is used for any of the following:
- Diagnosis, prevention, monitoring, treatment or alleviation of disease, disability, or injury, but not for disability or injury prevention
- Investigation, replacement, or modification of an anatomical, physiological, or pathological process
- Providing data via in-vitro examination of samples derived from a human body
What Will Change?
Compared to the previous MDD directive, the EU MDR regulations are considerably more comprehensive. They detail the obligations of the economic operators, the revised CE marking process and the identification and traceability of the devices. They also list the primary responsibilities and conditions for notified bodies, introduce post-market surveillance of marketed devices, detail confidentiality and funding obligations, establish penalties, tackle the topic of data protection and lay out the rules for cooperation between member states. In total, there are 13 differences noted by The FDA Group:
- The new regulation is four times longer, and contains five more annexes than its predecessor, the Medical Device Directive (MDD).
- The word “safety” appears 290 times in the MDR. The MDD, by comparison, uses it only 40 times.
- Significant changes in wording used in the new law will require companies to rationalize their portfolios and perform a global impact assessment in order to implement the necessary changes to remain compliant.
- Annex I, General Safety and Performance Requirements, identifies new conditions that will need to be addressed for most legacy devices (CE marked under the MDD). Existing products must be re-certified in accordance with the new regulations.
- The new rules will require most companies to update clinical data, technical documentation and labeling.
- Unique Device Identification (UDI) will be implemented to help track devices throughout the economic operator supply chain and will be required on all labels.
- While the scope of the MDD did not encompass medical purpose devices and AIMD, these are both included under MDR.
- The definition of medical device will be broadened to include non-medical and cosmetic devices not previously regulated. Examples include products for cleaning, disinfection or sterilization of devices as well as contact lenses, liposuction equipment or epilation lasers.
- Manufacturers will need to generate and provide more in-depth clinical data to prove safety and performance claims including tighter equivalency standards.
- Manufacturers will need to report all incidents, injuries and deaths into an EU portal (EUDAMED) that will centralize relevant data so that patients have access to more safety-related information. Reporting for incidents that did not result in death or serious deterioration in health is moved to 15 days from 30 days.
- Companies undergoing transition will need to revisit core processes including the quality assurance, risk management and post-market expectations. These will require careful review, planning and updating to re-implement in compliance with new requirements.
- Reclassification of many medical devices to a higher risk class and a new classification for reusable surgical devices requiring notified body oversight.
- IVDs are now classified into four risk classes that will require notified body review for about 90% of the devices, up from the current 10%.
As noted earlier, the MDR is focused on a lifecycle approach. Like the FDA, the EMA is not looking at the pure regulatory compliance of the device, but the quality of the product over its lifecycle. Manufacturers should have an integrated approach to design, production, quality assurance and that risk is being assessed throughout those phases of development and manufacture. In addition, they need a very robust change management system in place to know that if product changes are being made in one location, they are appropriately conveyed to other locations.
Article 10 (9) Manufacturers … shall establish, document, implement, maintain, keep up to date and continually improve a QMS that ensure(s) compliance… in the most effective manner and… that is proportionate to the risk class and… type of device.
Why is Post-Market Surveillance so Important?
Once a device has been approved by regulators and used by patients, there is an understanding that those products are going to be used in different patient populations than where they were originally tested. The purpose of post-market surveillance and establishing the Unique Device Identifier (UDI) is to understand how devices are performing in the market with patients. With the new EU MDR and iVDR, there is more responsibility on individual manufacturers to have robust systems and processes in place. Rather than just waiting for complaints and dealing with them as one-off issues, organizations need to have a proactive, systemic and a continuous improvement system in place. The global EUDAMED database will be on-line by May 26, 2022 in support of the new EU MDR and iVDR regulations and will be used to understand how products are performing in relation to similar technologies or to other ways in which that product could be used, and then feeding that back into the organization and conveying information systemically.
Are Medical Device Manufacturers Ready?
According to a research report, 70% of organizations are positive about meeting the MDR deadline.
“While it’s a positive sign that organizations feel optimistic about meeting their MDR and IVDR commitments, there’s clearly a lot of work to do post-pandemic,” explains Jon Hart, President of RWS Regulated Industries. “The decisions that medical manufacturing organizations make now won’t just affect their ability to comply with these two regulations, they will also play a continuing role in how quickly and effectively they can meet compliance standards (evolutions) in years to come. Introducing automation into the content management and change management process is a good first step, but a more visionary end-to-end solution that spans the manufacturing organization and the supply chain will better prepare organizations for the inevitable introduction of future regulations.”
Learn how you can better prepare your life sciences and medical device organizations for new regulations by embarking on your journey to becoming an Adaptive Manufacturing Enterprise.



